Définition
La maladie de Degos, également connue sous le nom de papulose atrophique, est une vasculopathie oblitérante rare, caractérisée par des lésions cutanées pathognomoniques. Décrite pour la première fois dans les années 1940 par Köhlmeier et Degos, cette maladie affecte principalement les jeunes adultes. Elle existe sous deux formes : une forme bénigne, limitée à la peau, et une forme maligne, multisystémique, souvent mortelle (papulomatose atrophiante maligne).
Épidémiologie
- Moins de 300 cas rapportés dans la littérature.
- Principalement de jeunes adultes.
- La maladie touche les deux sexes, mais des cas familiaux suggèrent une possible transmission génétique.
Physiopathologie
- Encore mal définie
- Prédisposition génétique ?
- Transmission autosomique dominante : certains cas familiaux soutiennent cette hypothèse
- Transmission liée à l’X : observée dans des familles où les hommes présentent une forme plus sévère de la maladie
- Des infections et des réactions auto-immunes pourraient jouer un rôle dans le développement de la maladie
- Trois mécanismes principaux sont impliqués dans sa pathogenèse :
- Vasculopathie
- Coagulopathie
- Dysfonction endothéliale
Clinique
Les manifestations cliniques de la maladie de Degos sont dominées par des lésions cutanées typiques et, dans sa forme maligne, par des atteintes systémiques :
- Lésions cutanées : présence de papules avec un centre blanc porcelaine déprimé et un bord érythémateux. Initialement petites, ces papules peuvent croître et devenir plus proéminentes avec le temps.
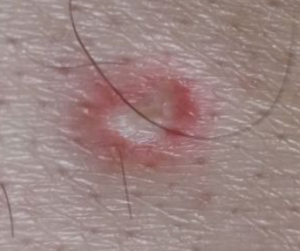
- Atteintes systémiques (forme maligne):
- Gastro-intestinales : Infarctus intestinal, perforation et péritonite.
- Neurologiques : Infarctus cérébraux, hémorragies sous-durales, méningites, myélite et calcifications du cortex.
- Cardiaques et pulmonaires : Péricardite constrictive, infarctus myocardique, atteinte de l’endocarde et pleurésie.
- Rénales : Épaississement des artérioles glomérulaires et néphropathie glomérulaire minime avec syndrome néphrotique.
Diagnostic
Le diagnostic de la maladie de Degos repose sur :
- Biopsie cutanée : Confirmation par l’observation de lésions typiques avec dépôts de C5b-9 et autres anomalies caractéristiques en histologie
- Imagerie : Pour détecter les atteintes systémiques
Évolution
L’évolution de la maladie de Degos peut varier :
- Forme bénigne : Restreinte à la peau, sans atteinte systémique.
- Forme maligne : Progrès vers des complications multisystémiques, avec une mortalité élevée (plus de 65 % dans certaines séries) principalement due aux atteintes gastro-intestinales et aux infarctus du système nerveux central.
Traitement
Le traitement de la maladie de Degos est principalement empirique et inclut :
- Antiagrégants plaquettaires et anticoagulants : Pour gérer la coagulopathie.
- Immunosuppresseurs et corticoïdes : Pour contrôler l’inflammation.
- Eculizumab : Inhibiteur du complément, efficace particulièrement pour la forme maligne avec atteinte gastro-intestinale, souvent combiné avec l’Iloprost pour prévenir les rechutes.
Perspectives thérapeutiques
Des traitements innovants offrent de nouvelles perspectives très prometteuses pour le contrôle de l’activité de la maladie :
- Inhibiteurs de l’interféron (anifrolumab) : Ciblant le récepteur IFNAR1.
- Inhibiteurs JAK : Bloquant l’activation en aval de l’interféron, pouvant réduire les complications en ciblant les voies spécifiques impliquées dans la pathogenèse de la maladie.